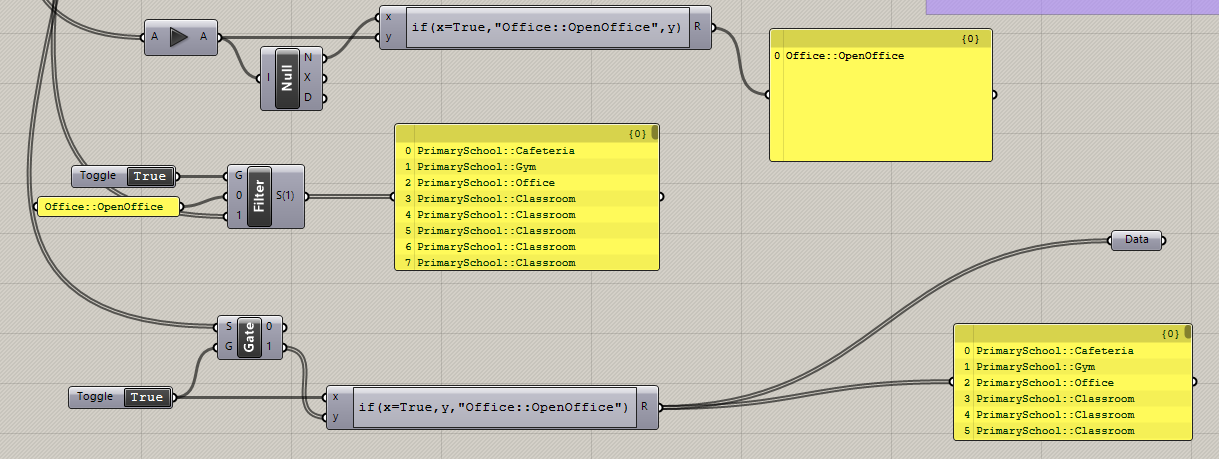
I have a large set of zone inputs that after each input is selected, the script takes a few seconds to compute…in an effort to save time I’d like to wait to send the inputs until all of them are filled in (it’s easy at they all combine into 1 panel into a Honeybee componenet)…I originally thought that a simple stream filter or stream gate would work, but for some reason it’s as if both those components “leak” data even when the gate is closed as the script is still taking some time to re-compute after each input change…data dam works perfect, however it’s a manual component and in a perfect world I’d like it to be Boolean controlled.
I’m sure I’m just doing something incorrect, so any insight would be appreciated!